
Research
Our current research focus on the development of new methodologies for selective organic transformations, with a particular emphasis on organocatalysis.The group efforts center on design and synthesis of novel homogeneous catalysts for achieving superior site- and stereoselectivity.

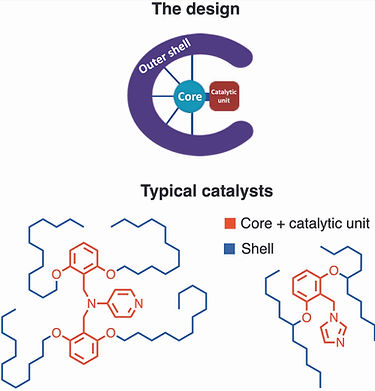
Core-shell nucleophilic organocatalysts for site-selective transformations
In the context of our efforts to develop site-selective modifications of amphiphilic diol and polyol molecules, we designed a series of core-shell organocatalysts, featuring a nitrogen-based Lewis base core enveloped by a secondary catalytic sphere. Both components of these catalysts shape their reactivity and site selectivity.
We employed N-alkylimidazole and p-aminopyridine core units, though other nucleophilic aza-heterocycles are under consideration. In general, p-aminopyridine-based catalysts are much more active and moderately more selective than those based on imidazole.
The secondary sphere modulates the selectivity and activity of the core through direct electronic influence and through-space non-covalent interactions. In apolar media, the influence of the lipophilic secondary sphere is ambivalent in terms of activity and noticeably augments the site selectivity, favoring the apolar domain of amphiphiles. Conversely, under similar conditions, an oligoethylene glycol-based secondary sphere generally enhances catalyst activity – sometimes dramatically. Its influence on site selectivity is reaction-dependent, enhancing selectivity in phosphorylation while reducing it in acylation.
We are currently exploring additional core-shell combinations and secondary sphere components to further enhance site selectivity in the reactions of interest.
Chiral core-shell organocatalysts
Our studies on achiral core-shell nitrogen-based organocatalysts have revealed that the steric and electronic properties of the outer sphere strongly affect the course of the reactions they catalyze, particularly their selectivity. This transfer of information from the catalyst’s outer sphere to the reacting substrates, likely mediated by non-covalent interactions such as dispersion forces and n–cation electron donation, shapes site selectivity in the examined transformations.
Building on this understanding, we postulate that similar interactions could also play a crucial role in conveying chirality from a chiral catalytically active core via the outer shell to the substrates. This process will direct their approach to the reactive site of the catalyst, plausibly inducing enantioselectivity in the catalytic transformations.
In line with this hypothesis, we design catalysts incorporating a chiral core, featuring an interlinked dibenzylaminopyridine catalytic moiety and decorated with the alkoxy outer sphere appendages, akin to those in some of the achiral catalysts. Our preliminary results support their potential for both enantio- and site-selectivity. Since the strongest outer sphere effects on site selectivity were demonstrated by the progenitor achiral catalysts in alcohol phosphorylation, we are applying the newly devised chiral catalysts to enantioselective versions of this transformation, a notably underexplored methodology.
To broaden the set of potential catalysts, chiral cores based both on point and axial chirality are being used. For the same reason, both hydrocarbon- and oligoether-based outer sphere appendages (both showing a promising influence in site-selective phosphorylations) are employed in the catalyst design.
Site-selective transformations of natural products
Building on the extensive expertise our group has acquired over the past few years in the site-selective modification of amphiphilic diols using model substrates, we now aim to establish methodology for site-selective functionalization of diol and polyol natural products possessing amphiphilic character. Our current focus is on achieving selective acylation and phosphorylation of such substrates.
For instance, we recently succeeded in reversing the native acylation preference of midecamycin-A1, a macrolide antibiotic from the leucomycin family that features distinct polar and apolar domains. Typically, acylation occurs preferentially at the alcohol located in the polar region. However, by combining an optimized catalyst with a more reactive acylating agent, we redirected the reaction to favor acylation at the alcohol within the apolar domain.
Other modifications of this complex substrate, as well as other naturally occurring di- and polyols that combine both polar and apolar domains (e.g., baccatin III, tacrolimus), are currently being explored. Site-selective methodologies tailored for complex multifunctional substrates are pivotal to the late-stage modification approach, which has emerged in recent years as a powerful and transformative strategy for the preparation of bioactive molecules.
